

Abstract
INTRODUCTION. Calreticulin (CALR) is mutated in 20% of pts with essential thrombocythemia (ET) and primary myelofibrosis (PMF). The most frequent mutations are a 52 bp deletion (T1) and a 5 bp insertion (T2) in exon 9, that cause a recurrent frameshift resulting in novel C-terminal sequence common to all mutant CALR proteins. Recent data indicate that interaction of mutant CALR with the thrombopoietin receptor MPL contributes to the abnormal megakaryocytopoiesis (Mk) of ET/PMF (Araki M et al & Chachoua I et al, Blood 2016; Elif S et al, Blood 2018), however full characterization of mechanisms pertaining to mutant CALR remains to be pursued.
METHODS. By using CRISPR/Cas9 editing, we generated CALR knock-out (KO) variants starting from cord blood (CB) CD34+ cells, K562, UT7 and HL-60 cell lines, and CALR T1 variants from K562 and UT7 cells. Stable expression of CALR wild-type (WT), T1 and T2 was obtained by viral transfection of K562-KO cells.
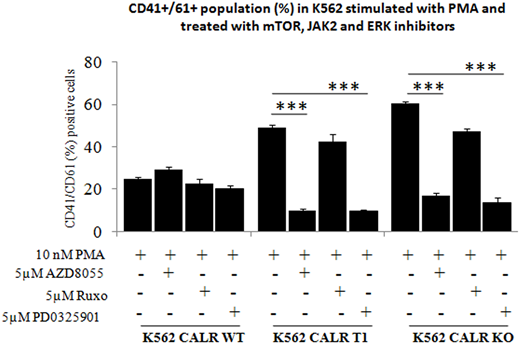
RESULTS. In the different genome-edited cell models, KO or T1 mutation did not result in appreciable changes in proliferation rate, cell cycle and apoptosis under standard culture conditions. However, UT7 KO and T1 cells were able to grow in the absence of GM-CSF, that was otherwise necessary for maintenance of WT counterpart, indicating cytokine independence similarly imparted by deletion or T1 mutation of CALR. To evaluate impact of mutated CALR on Mk commitment, we induced parental, KO and T1 K562 cells with phorbol-myristate acetate (PMA); at day 3 after induction, 60% and 48% of KO and T1 cells, respectively, expressed CD41/CD61 compared to 24% of parental cells (p<0.01). Similar findings were noted in KO K562 cells that were transfected with vectors encoding the WT, T1 and T2 mutated CALR: CD41/CD61+ cells increased from 30% in parental cells to 70%, 55%, 62%, respectively, in KO cells and cells expressing T1 and T2 mutant CALR (p<0.01). We also found that a greater proportion of KO and T1 UT7 cells, maintained in either GM-CSF and TPO, spontaneously expressed CD41/CD61 at d7 of culture (50-51% and 41-49%, respectively, for KO and T1 cells with GM-CSF or TPO compared to 26-25% of parental cells; p<0.01) and underwent morphologically recognizable Mk maturation. Mk clonogenic assays were initiated in plasma clot cultures by plating CB-derived CD34+ cells with CRISPR/Cas9-induced deletion of CALR (KO cells). KO CD34+ cells generated a number of CFU-Mk colonies that was 10-fold higher than untouched CD34+ cells (p<0.05). Overall, these data suggested that deletion of CALR results in promotion of Mk differentiation in cell lines and primary cells thereby mimicking the effects of CALR T1. Array phosphoproteomic assay in K562 KO cells showed phosphorylation levels of p38γ, p70S6k mTOR, Erk1 and Erk2 from 0.5 to >2-fold higher compared to parental cells. Then, we induced KO and T1 K562 cells to Mk differentiation in the presence of JAK2, mTOR and Erk1/2 -inhibitors; only mTOR and Erk1/2 -inhibitors significantly (P< 0.05) reduced Mk differentiation of T1 and KO cells by respectively 5- and 4- fold with mTOR inhibitor and 5- and 4.5- fold with Erk1/2 inhibitor compared to parental cells, that were largely insensitive to inhibitors (Figure).
Isolated megakaryocytes from CALR-mutant patients were found to present abnormally increased release of Ca2+ from endoplasmic reticulum (ER) compared to control cells (Pietra D et al. Leukemia, 2016). We similarly found that both KO and T1 K562 cells presented impaired capability in restoring calcium homeostasis compared to parental cells under treatment with ionophores thapsigargin and ionomycin. Finally, we comparatively assessed MPO expression by flow cytometric analysis in parental and CALR KO HL-60 cell lines; increased myeloperoxidase (MPO) degradation was previously reported in MPN pts expressing CALR mutation (Theocharides A et al., Blood, 2016). We found significant reduction of MPO levels (9-fold lower, p<0.01) in KO compared to parental cells, while MPO mRNA levels were not affected by CALR KO, suggesting similarly enhanced degradation of MPO in cells with targeted deletion of CALR.
CONCLUSIONS. Overall, our data suggest that the novel C-terminus of CALR originating from exon 9 mutations loses some physiologically functions that are phenocopied by absence of CALR protein in cells made KO for the gene in a MPL-independent context.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal